
M. Neal Waxham, PhD
- Professor
- William M. Wheless III, Professor in Biomedical Sciences
Biography
Molecules Responsible For The Formulation And Storage Of Memories
Our memories are formed and stored in cells within the nervous system. How these memories are encoded and retained throughout an individual’s lifetime is the fundamental question being investigated in this laboratory. While we are concerned with determining how neurons communicate by sending chemical signals across synapses, we are even more interested in determining how such communication changes with experience — a process commonly referred to as synaptic plasticity. Identification of the mechanisms responsible for these changes will be significant in our attempts to understand the formation and storage of memories.
Increased intracellular Ca2+, activation of calmodulin and activation of Ca2+/calmodulin-dependent protein kinases, and other enzymes form a biochemical pathway that can lead to short-term and long-term changes in synapse structure and function. However, few attempts have been made to experimentally address an integrated cellular model that encompasses when, where and how activation of each of these components leads to functional alterations. We are in the process of characterizing each step in this pathway utilizing biochemical, molecular biological and optical techniques applied to both in vitro and in situ models.
Our goals necessitate understanding the cellular distribution and limits of diffusion of each molecule in this pathway under experimental conditions that would lead to functional changes like synaptic plasticity. To address this issue, neurons are injected with fluorescently labeled molecules and their distribution and rates of diffusion are monitored with optical imaging. Multi-channel confocal microscopy also provides us with a high-resolution understanding of the relative distribution of each molecule within neurons.
Additional studies utilizing molecular, biochemical and immunocytochemical techniques are attempting to discover how calmodulin and calmodulin-dependent enzymes associate with other intracellular proteins to attain their appropriate intracellular localization. At the molecular level, we must understand how calmodulin interacts with target enzymes and what physiological factors can regulate that association. To approach this question we express calmodulin and calmodulin-dependent enzymes, mutated at defined sites, purify and fluorescently label them, and then apply fluorescent techniques to monitor rates of association and dissociation. We hope to consolidate results from each of these studies to provide an integrated picture of how synaptic plasticity occurs at the molecular and cellular level.
Areas of Interest
Research Interests
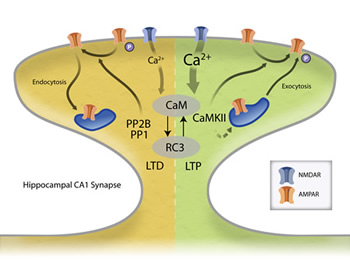
Long-term potentiation (LTP) and long-term depressions (LTD) at cortical synapses both require Ca2+ influx through NMDA receptors in the post synaptic cell and the activation of calmodulin (CaM). Importantly, other proteins at synapses (like RC3) can bind to CaM and alter its Ca2+-binding properties. Through unknown mechanisms, Ca2+/CaM then activate either CaM-dependent protein phosphatase (PP2B also known as calcineurin) or Ca2+/CaM-dependent protein kinase II (CaMKII). Depending on this choice, the synapse either gets weaker (LTD) or stronger (LTP). Once activated, PP2B and CaMKII can influence the physiological properties of the synapse by phosphorylating/dephosphorylating the AMPA subtype of glutamate receptors to change their properties or influence AMPA receptor number by affecting protein trafficking. We are investigating how the Ca2+ signal is decoded by this biochemical network and how these activated enzymes lead to long-term plasticity.
Publications
- Heberle, FA., Doktrova, M., Scott, H.L., Skinkle, A.D., Waxham, M.N., and Levental, I. (accepted, in press). Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proc. Natl. Acad. Sci. USA
- Symons, J., Cho, K.-J., Chang, J., Du, G., Waxham, M.N., Hancock, J.F., Levental, I. and Levental, K.R. (2020). Lipidomic atlas of mammalian cell membranes reveals hierarchical variation induced by culture conditions, subcellular membranes, and cell lineages. Soft Matter. DOI: 10.1039/D0SM00404A.
- Maynard, M.E., Redell, J., Kobori, N., Underwood, E., Fischer, T.D., Hood, K.N., LaRoche, V., M N Waxham, M.N., Moore, A.N. and Dash, P.D. (2020). Loss of PTEN-induced kinase1 (Pink1) reduces hippocampal tyrosine hydroxylase and impairs learning and memory. Exp. Neurol. Jan;323:113081. doi: 10.1016/j.expneurol.2019.113081.
- Ezerski, J.C., Zhang, P., Jennings, N.C., Waxham, M.N. and Cheung, M.S. (2020). Molecular Dynamics Ensemble Refinement of Intrinsically Disordered Peptides According to Deconvoluted Spectra from Circular Dichroism. Biophys J. 2020 Feb 25. pii: S0006-3495(20)30164-8. doi: 10.1016/j.bpj.2020.02.015.
- Liman, J., Bueno, C., Eliaz, Y., Schafer, N.P., Waxham, M.N., Wolynes, P.G., Levine, H., and Cheung, M.S. (2020) The role of the Arp2/3 complex in shaping the dynamics and structure of branched actomyosin networks. Proc. Natl. Acad. Sci. USA.
- Wang,Q., Chen, M., Schafer, N.P., Bueno, C. Song, S.S., Hudmon, A. Wolynes, P.G., Waxham, M.N. and Cheung, M.S. (2019) Calcium/calmodulin-dependent kinase II – Actin assemblies and their dynamic regulation by calmodulin in dendritic spines. Proc. Natl. Acad. Sci. USA.
- Scott, H., Skinkle, E., Kelley, E., Waxham, M.N., Levental, I. and Heberle, F.A., (2019) On the mechanism of bilayer separation by extrusion; or, why your large unilamellar vesicles are not really unilamellar. Biophysical J.
- Gireud-Goss, M., Reyes, S., Wilson, M., Farley, M., Memarzadeh, K., Srinivasan, S., Sirisaengtaksin, N., Yamashita, S., Tsunoda, S., Lang, F.F., Waxham, M.N., Bean, A.J. (2018) Distinct mechanisms enable inward or outward budding from late endosomes/multivesicular bodies. Exp. Cell Res. 372, 1-15. https://www.sciencedirect.com/science/article/pii/S001448271830778X?via%3Dihub
- Fischer, D., Dash, P.K., Liu, J. and Waxham, M.N. (2018) Morphology of Mitochondria in Spatially Restricted Axons Revealed by Cryo-Electron Tomography. PloS Biology. e2006169. https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.2006169
- Park, D., Hu, B., Lara-Tejero, M., Waxham, M.N., Galan, J. and Liu, J. (2018) Visualization of the type III secretion mediated Salmonella-host cell interface using cryo-electron tomography. eLife. e39514. https://elifesciences.org/articles/39514