
Peter Doris, PhD
- Professor & Director, IMM-Center for Human Genetics
- Professor and Mary Elizabeth Holdsworth Distinguished University Chair in Metabolic and Inflammatory Disease Research
Biography
Dr. Doris received his doctoral degree at the University of California and returned to his native England as a Medical Research Council post-doctoral fellow at Cambridge University and the University of Reading. He joined the Institute in the summer of 1997 and is appointed as Mary Elizabeth Holdsworth Distinguished University Chair and Professor of Molecular Medicine in the Center for Human Genetics. His research focuses on mechanisms of common chronic cardiovascular disease including hypertension and the injury that it causes to the kidney and brain.
Research Profile:
Out-living our kidneys: Our kidneys occupy a rough neighborhood in our bodies. Although they receive a sizable fraction of the oxygenated blood our hearts pump out, they are constantly on the verge of oxygen deficiency. Indeed, this characteristic has been adapted as a “feature” rather than a “bug” by evolution: it is our kidneys that are used as a monitor whether we need to increase the number of oxygen-carrying red cells in our blood. Our kidneys control the production of red cells by the bone marrow and determine the oxygen-carrying capacity of the blood. Since the kidneys are also our excretory organs, their neighborhood is made tougher by the continuous filtration and concentration of waste products that must be eliminated from our blood. It is not so surprising then, that as we age, our kidney function declines. As medicine advances and we live longer lives, a growing portion of society is outliving their kidneys and face long-term renal dialysis or transplant for survival. We seek to understand what causes progressive loss of renal function and how is this process amplified by high blood pressure.
Genetics and kidney function: The best predictor of whether an individual will lose enough kidney function to require dialysis is whether they have a first or second degree relative who has reached this “end stage” of renal disease. Aggregation of kidney disease within families indicates that inherited factors shape risk of renal disease. At present, the mechanism of renal functional decline in most people is not known. Kidneys are difficult to study in humans because they lie deep within the body and their functional units, the glomeruli and nephrons, are microscopic structures. Increased blood pressure is a major risk factor that amplifies the age-related decline in renal function. We study this disease process of increased blood pressure (hypertension) and loss of kidney function in a rodent genetic model. In this model, the genetic factors that influence both blood pressure and kidney disease are separate – elevated blood pressure is a pre-condition necessary for the other genetic risk factors to create disease. We are working to identify the genes involved and the pathways that are disturbed by effects arising in these genes.
Progressive renal disease and immune function: Our recent work reveals that genetic variation in each of two major immune system genes combine to play a key role in susceptibility to injury. Blood pressure elevation exceeds the ability of the kidney to match blood flow to the local needs for oxygen and nutrients in the kidney tissue. This triggers an inflammatory process in which immune responses are provoked by damage to the tissue. Genetic variation appears to determine whether the outcome is injury and repair or injury leading to sustained inflammation and further tissue damage. We can change the course of disease by drugs that modify the immune response. Now have identified immune genes that shape the immune response, our immediate goal is to understand which immune cells are expressing these genes and which aspects of their function are altered so as to create a self-damaging immune response. With this information in hand, we will be able to seek and narrowly target therapeutic approaches that can sustain kidney function while allowing the normal protective functions of the immune system to be preserved.
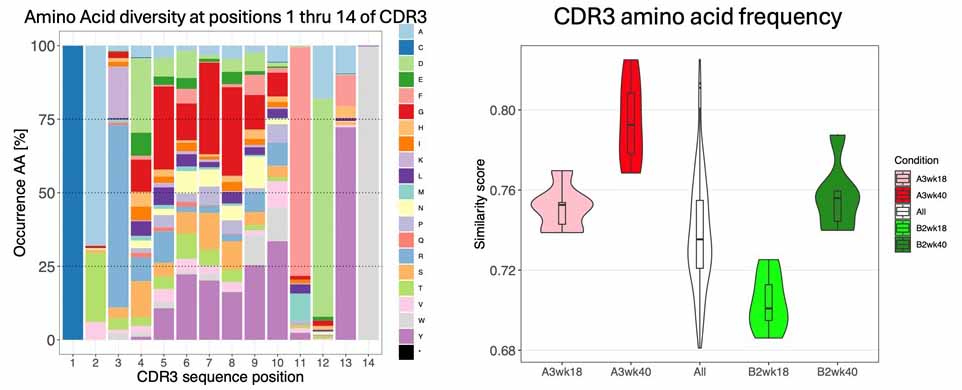
Figure 1. Left: the most important part of an antibody molecule for antigen recognition is the CDR3 region. This part of the antibody is “evolved” by the host to make antibodies specific for a pathogen. This occurs by intentional mutation events that change the amino acid composition and sequence of the CDR3. The colors represent individual amino acids and between the first and last (positions 2-13) where a wide range of animo acids are “evolved” to target a wide range of antigens. Right: We can estimate how diverse the antibodies produced are and how this can be affected by inheritance of antibody genetic variation. The pink distribution shows that, at 18 weeks of age, the A3 genome has a less diverse (more similar) antibody response than the genome of the light green (B2) strain. As the animals age, antibody responses become more stereotyped and diversity declines at 40 weeks. But it declines more in the red (A3 genome) than in the green (B2 genome). This means that the antibodies we are able to make is shaped by inheritance.
Heritability of antibody responses: Our work implicates genetic variation in the part of the genome from which antibody genes are expressed. Indeed, we have had to focus much effort on unravelling the enormous extent of genetic variation in this region. It is easy to imagine that the genes involved in such life-saving protection from pathogens have to evolve rapidly in order to adjust to the even more rapid evolution of pathogens. Another feature of such life-saving genes is that the variation that occurs in them often involves a trade-off between increase protection from pathogens and potentially harmful effects (autoimmunity) on the host of the antibodies. Our studies of heritability of antibody responses indicate that the genomic region involved changes over time principally by duplication and deletion events. We have shown that this can affect how much antibody is generated from the genome creating high and low antibody producers. We have also shown that the actual antibodies made are shaped by the genetic variation (Figure 1) so that this variation affects the profile of antibody responses.
Publications
- T.S., Smith, M.L., Ciosek, J.L., Li, K., Doris, P.A. Three decades of rat genomics: approaching the finish(ed) line. Physiological Genomics PMID:39348459.
- Li, K., Smith, M.L., Blazier, J.C., Kochan, K.J., Wood, J, Howe, K.J., Kwitek, A.E., Dwinell, M., Chen, H., Ciosek, J., Murphy, T.D., Kalbfleisch.T.S., Doris, P.A. GRCr8: construction and evaluation of new rat reference genome assembly from long reads and long-range scaffolding. Genome Research. In Press.
- T V de Jong, Y Pan, P Rastas, Dl Munro, M Tutaj, H Akil, C Benner, A S Chitre, W Chow, V Colonna, C L Dalgard, W M Demos, P A Doris, E Garrison, A Geurts, H M Gunturkun, Vi Guryev, T Hourlier, K Howe, J Huang, T Kalbfleisch, P Kim, L Li, S Mahaffey, F J Martin, P Mohammadi, A B Ozel, O Polesskaya, M Pravenec, P Prins, J Sebat, J R Smith, L C Solberg Woods, B Tabakoff, A Tracey, M Uliano-Silva, F Villani, H Wang, B M Sharp, F Telese, Z Jiang, L Saba, X Wang, T D Murphy, A A Palmer, A E Kwitek, M R Dwinell, R W Williams, J Z Li, H. Chen A revamped rat reference genome improves the discovery of genetic diversity in laboratory rats. Cell Genomics, 4(4):100527, 2024 PMID:38537634
- Dhande, I, Y. Zhu, A. S. Joshi, M.L. Smith, T.S. Kalbfleisch, J. Hicks, M.C. Braun, P. A. Doris. Polygenic genetic variation affecting antibody formation underlies renal injury in the Stroke-Prone Spontaneously Hypertensive Rat. Am. J.Physiol (Renal). 325(3):F317-F327, 2023. PMID:37439198
- T.S., Hussien, N.A., Li, K., Brashear, W.A., Kochan, K.J., Hillhouse, A.E., Zhu, Y., Dhande, I.S., Hudson, E.A., Sumlut, M.H., Murphy, T., Thibaud-Nissen , F., Smith, M.L., Doris, P.A. The assembled genome of the stroke-prone spontaneously hypertensive rat. Hypertension. 80(1):138-146. 2023 PMID:36330812.
- Saha, P., Mell, B., Golonka, R.M., Bovilla, V.R., Mei, X., Abokor, A.A., Yeoh, B.S., Doris, P.A., Gewirtz, A.T., Joe, B., and Vijay-Kumar, M. Immunoglobulin A is Insufficient and Quiescent in Spontaneously Hypertensive Rats. Hypertension, 79(10):2239-2249, Oct, 2022. PMID:35950503.
- De Jong, T, Chen, H, Brashear, W.A., Kochan, K.J., Hillhouse, A.E., Zhu, Y., Dhande, I.S., Hudson, E.A., Sumlut, M.H., Smith, M.L., Kalbfleisch.T.S., Doris, P.A. A new rat genome reference assembly: progress and problems. Physiological Genomics, 54:251 -260, 2022 PMID:35543507.
- Gray, E.A., S. Patel, A. Doris, and T. Hussain. Combinatory Approach Targeting AT2R and Neprilysin Provides Superior Reno-protection Compared to Entresto in Obese Rats. Frontiers in Pharmacol., 12:778953, 2022 PMID:35197849.
- Dhande, I.S., M.C. Braun and A. Doris. Emerging insights into chronic renal disease pathogenesis in hypertension from human and animal genomic studies. Hypertension, 78(6): 1689–1700, 2021 PMID 34757770
- Dhande, I.S., and A. Doris Genomics and inflammation in cardiovascular disease. Comprehensive Physiology, 11(4):1-22, 2021. PMID:34570903
- Dhande, I.S., and A. Doris. Pulling the hood off genetic susceptibility to hypertensive renal disease. J. Amer. Soc. Nephrol., 31(4):667-668, 2020. PMID 32123053
- Dhande, I.S., Y. Zhu, S.C. Kneedler, A.S. Joshi, M.J. Hicks, S.E. Wenderfer, M.C. Braun, A. Doris. Stim1 polymorphism disrupts immune signaling and creates renal injury in hypertension. J. Amer. Heart Association. 9(5):e0141422019, 2020. PMID 32075490
- Dhande, I.S., C. Kneedler, Y. Zhu, A.S. Joshi, M.J. Hicks, S.E. Wenderfer, M.C. Braun, P.A. Doris. Natural genetic variation in Stim1 creates stroke in the spontaneously hypertensive rat. Genes and Immunity. 2020. PMID 32300198