Molecular mechanisms underlying working memory and its dysfunction after TBI
Working memory is the process of actively maintaining and integrating information in mind for a relatively short period of time in order to solve a goal-directed task. Our investigations into the intracellular mechanisms of working memory led to the surprising discovery that protein kinase activity (e.g. CaMKII, PKA) in the medial prefrontal cortex impairs working memory, a mechanism opposite to that shown for short- and long-term memory formation. Instead, we have demonstrated that calcium-dependent phosphatases (that are known to impair short-term memory) are required for working memory. These findings led to the development of a model that the balance of phosphatase and kinase activity determines whether working memory is engaged or impaired in order to solve a goal-directed task (Figure 1). While direct damage to the prefrontal cortex can result in working memory dysfunction, impairments can also be observed in the absence of overt neuronal loss within this structure. Studies performed in humans, monkeys and rodents have shown that the neurotransmitter dopamine plays a critical role in working memory, with both excessive and insufficient dopamine receptor D1 stimulation impairing working memory performance. We have observed that TBI causes a rapid and sustained increase in prefrontal dopamine levels. This increase, via stimulation of post-synaptic D1 receptors on inhibitory neurons, increases the expression of glutamic acid decarboxylase (GAD), the rate-limiting enzyme in GABA synthesis. We discovered that this increase in GAD levels causes excessive GABAergic inhibition of prefrontal neurons, leading to working memory dysfunction. While these neurotransmitter imbalances cause working memory dysfunction early after injury, our results also show that working memory deficits at later time points are associated with morphological changes (e.g. reduced dendrite length and increased spine numbers) in the pyramidal neurons within the prefrontal cortex (Figure 2). Interestingly, we have discovered that morphological changes occur in the prefrontal cortex even after mild brain injury and that these changes are associated with deficits in prefrontal cortex-dependent behaviors.
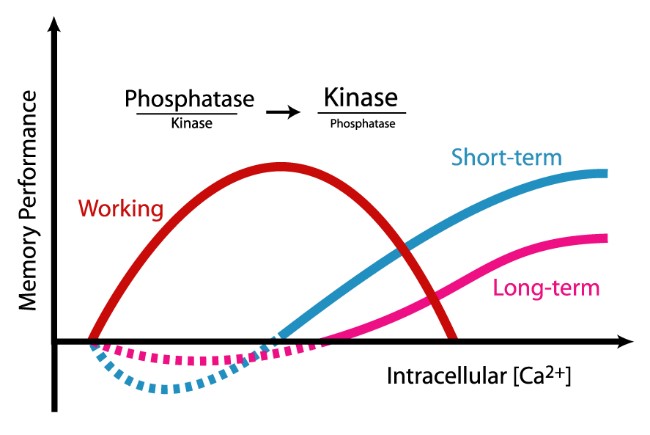 Figure 1. Research in our laboratory has determined that the molecular mechanisms that underlie working memory are antagonistic for short- and long-term memory. We have discovered that while short- and long-term memory requires activation of second messenger kinases, working memory requires phosphatase activity. This molecular switch may represent the means by which information held in working memory is not stored more long-term. Further, this research indicates that drugs which activate protein kinases to improve short- or long-term memory may have detrimental effects on working memory.
Figure 1. Research in our laboratory has determined that the molecular mechanisms that underlie working memory are antagonistic for short- and long-term memory. We have discovered that while short- and long-term memory requires activation of second messenger kinases, working memory requires phosphatase activity. This molecular switch may represent the means by which information held in working memory is not stored more long-term. Further, this research indicates that drugs which activate protein kinases to improve short- or long-term memory may have detrimental effects on working memory.
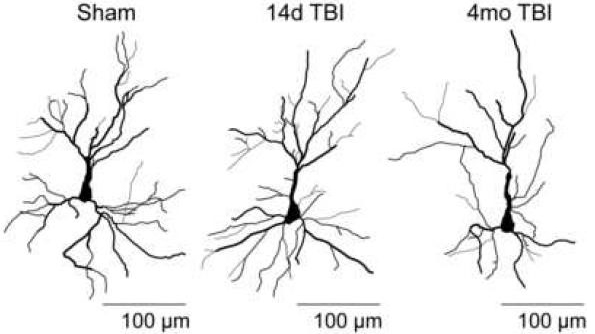 Figure 2. Traumatic brain injury (TBI) can cause working memory dysfunction in the absence of neuronal death in the prefrontal cortex. Our laboratory has identified several cellular and molecular events that contribute to working memory dysfunction, including morphological changes of neurons in the prefrontal cortex. In addition to causing working memory dysfunction, our results implicate these changes in other prefrontal cortex-dependent behaviors such as fear memory extinction. These findings may provide a cellular explanation for the link between brain injury and post-traumatic stress disorder (PTSD).
Figure 2. Traumatic brain injury (TBI) can cause working memory dysfunction in the absence of neuronal death in the prefrontal cortex. Our laboratory has identified several cellular and molecular events that contribute to working memory dysfunction, including morphological changes of neurons in the prefrontal cortex. In addition to causing working memory dysfunction, our results implicate these changes in other prefrontal cortex-dependent behaviors such as fear memory extinction. These findings may provide a cellular explanation for the link between brain injury and post-traumatic stress disorder (PTSD).
Selected reading:
- Runyan J.D., Moore A.N., and Dash P.K. A Role for prefrontal calcium-sensitive protein phosphatase and kinase activities in working memory. Learning and Memory. 12:103-110, 2005.
- Runyan J.D. and Dash P.K. Distinct prefrontal molecular mechanisms for information storage lasting seconds versus minutes. Learning and Memory. 12:232-238, 2005.
- Kobori, N., Clifton, G.L., Dash, P.K. Enhanced catecholamine synthesis in the prefrontal cortex following traumatic brain injury: Implication for prefrontal dysfunction. J. Neurotrauma, 23:1094-1102, 2006
- Kobori, N., and Dash, P.K. Reversal of brain injury-induced prefrontal GAD expression and working memory deficits by D1 receptor antagonism. J. Neuroscience. 26:4236-4246, 2006
- Dash, PK, Moore, AN, Kobori, N and Runyan, RD. Molecular activity underlying working memory. Learning and Memory 14: 554-563, 2007.
- Hoskison, M.M., Moore, A.N., Hu, B., Orsi, S.A., Kobori, N. and Dash, P.K. Persistent working memory dysfunction following traumatic brain injury: evidence for a time-dependent mechanism. Neuroscience, 159:483-491, 2009.
- Kobori, N., Hu, B. and Dash, P.K. Altered adrenergic receptor signaling following traumatic brain injury contributes to working memory dysfunction. Neuroscience, 172: 293-302, 2011.
- Zhao J, Huynh J, Hylin MJ, O’Malley JJ, Perez A, Moore AN, Dash PK. Mild Traumatic Brain Injury Reduces Spine Density of Projection Neurons in the Medial Prefrontal Cortex and Impairs Extinction of Contextual Fear Memory. J Neurotrauma. 35:149-156, 2018