Cellular and molecular mechanisms underlying neuronal death, and learning and memory impairments following TBI
Traumatic brain injury (TBI) is a major worldwide health concern. Memory impairments profoundly compromise independent living and quality of life for people who have sustained a TBI. Furthermore, TBI (also repeated mild TBI or concussion) has been established a risk factor for neurodegenerative diseases such as Parkinson’s disease (PD), Alzheimer’s disease (AD) and chronic traumatic encephalopathy (CTE). Currently, there are no effective treatments to reduce brain damage and improve learning and memory for TBI patients. Our multi-disciplinary approach has discovered that activating endogenous cytoprotective mechanisms, reducing inflammation, enhancing energy production (Figure 1), and limiting ER stress (Figure 2) in the acute phase can offer neuroprotection and attenuate memory impairments. Furthermore, we have also shown that epigenetic regulation by microRNAs, both in experimental and clinical studies, may contribute to TBI pathology and outcome. As TBI activates multiple pathological cascades, it is likely that a combination therapy targeting these mechanisms would be more successful as a treatment.
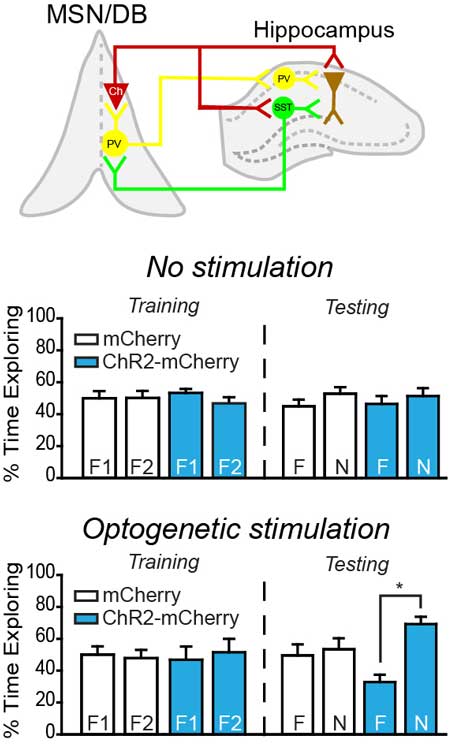 Figure 1. The hippocampus plays a prominent role in learning and memory formation. The functional integrity of this structure is often compromised following traumatic brain injury (TBI), resulting in lasting cognitive dysfunction. The activity of hippocampal neurons, particularly place cells, is coordinated by local theta oscillations. Hippocampal theta oscillations are generated in the medial septal nucleus/diagonal band (MSN/DB) and are the result of modulation within the hippocampus by cholinergic neurons and parvalbumin-positive inhibitory neurons. We have found that hippocampal theta oscillations are reduced as a result of mild TBI and are associated with poor performance in hippocampus-dependent learning and memory tasks. Using an optogenetic stimulation approach, we have found that the memory impairments observed in brain injured animals in the absence of stimulation can be reversed by optogenetically stimulating CA1 pyramidal neurons (expressing ChR2-mCherry) at theta during learning. These results suggest that direct stimulation of CA1 pyramidal neurons at theta may be a viable option for enhancing memory following TBI.
Figure 1. The hippocampus plays a prominent role in learning and memory formation. The functional integrity of this structure is often compromised following traumatic brain injury (TBI), resulting in lasting cognitive dysfunction. The activity of hippocampal neurons, particularly place cells, is coordinated by local theta oscillations. Hippocampal theta oscillations are generated in the medial septal nucleus/diagonal band (MSN/DB) and are the result of modulation within the hippocampus by cholinergic neurons and parvalbumin-positive inhibitory neurons. We have found that hippocampal theta oscillations are reduced as a result of mild TBI and are associated with poor performance in hippocampus-dependent learning and memory tasks. Using an optogenetic stimulation approach, we have found that the memory impairments observed in brain injured animals in the absence of stimulation can be reversed by optogenetically stimulating CA1 pyramidal neurons (expressing ChR2-mCherry) at theta during learning. These results suggest that direct stimulation of CA1 pyramidal neurons at theta may be a viable option for enhancing memory following TBI.
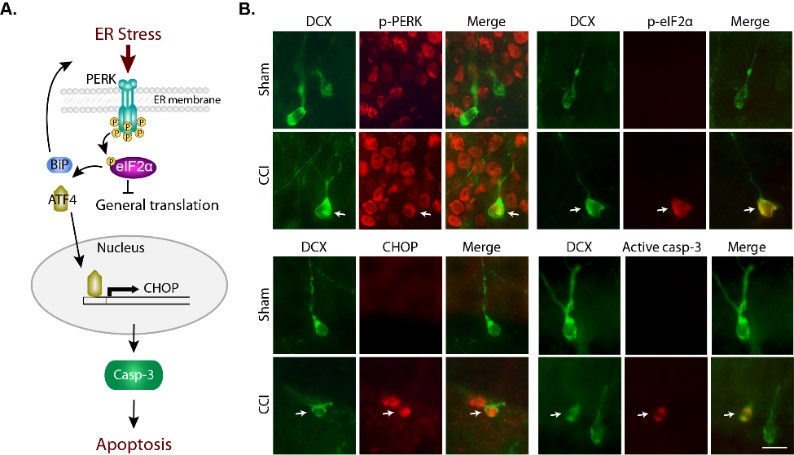 Figure 2. Endoplasmic reticulum (ER) stress results in the production of misfolded proteins. A key cellular pathway that serves to resolve ER stress is the PERK-eIF2α pathway, which blocks protein synthesis. However, if the stress is intense and cannot be resolved, the cell can die via CHOP-mediated apoptosis. We have discovered that traumatic brain injury results in the apoptotic death of neurons, and that this death is mediated in part by activation of the CHOP cascade. Blockade of CHOP, or facilitation of stress resolution, results in recued neuronal loss and improved learning and memory after TBI.
Figure 2. Endoplasmic reticulum (ER) stress results in the production of misfolded proteins. A key cellular pathway that serves to resolve ER stress is the PERK-eIF2α pathway, which blocks protein synthesis. However, if the stress is intense and cannot be resolved, the cell can die via CHOP-mediated apoptosis. We have discovered that traumatic brain injury results in the apoptotic death of neurons, and that this death is mediated in part by activation of the CHOP cascade. Blockade of CHOP, or facilitation of stress resolution, results in recued neuronal loss and improved learning and memory after TBI.
Selected Readings
- Zhao, J., Moore, A.N., Redell, J.B. and Dash, P.K.: Enhancing expression of Nrf2-driven genes protects the blood-brain barrier following brain injury. J. Neurosci. 19: 10240-10248, 2007.
- Redell JB, Moore AN, Ward NH 3rd, Hergenroeder GW, Dash PK. Human traumatic brain injury alters plasma microRNA levels. J Neurotrauma. 27:2147-2156, 2010.
- Menge T, Zhao Y, Zhao J, Wataha K, Gerber M, Zhang J, Letourneau P, Redell J, Shen L, Wang J, Peng Z, Kozar R, Cox CS, Khakoo AY, Holcomb JB, Dash PK and Pati S. Mesenchymal stem cells regulate blood-brain barrier integrity in traumatic brain injury through production of the soluble factor TIMP3. Science Trans. Med. 4:161ra150, 2012.
- Dash PK, Zhao J, Kobori N, Redell JB, Hylin MJ, Hood, KN and Moore AN. Activation of alpha7 cholinergic nicotinic receptors reduces blood-brain barrier permeability following experimental traumatic brain injury. J. Neurosci, 36:2109-2818, 2016.
- Fischer T, Hylin MJ, Zhao J, Moore, AN, Waxham MN and Dash PK. Altered mitochondrial dynamics and TBI pathophysiology. Front. Systems Neurosci. 10:29, 2016.
- Zhao J, Hylin MJ, Kobori N, Hood KN, Moore AN, Dash PK. Post-Injury Administration of Galantamine Reduces Traumatic Brain Injury Pathology and Improves Outcome. J Neurotrauma. 35:362-374, 2018.
- Hood KN, Zhao J, Redell JB, Hylin MJ, Harris B, Perez A, Moore AN and Dash PK. Endoplasmic reticulum stress contributes to the loss of newborn hippocampal neurons after traumatic brain injury. J. Neurosci. 38:2372-2384, 2018.