Soto Lab Research
Dr. Soto’s lab focuses on the investigation of the molecular basis of Protein Misfolding Disorders, mainly studying Alzheimer’s disease (AD) and prion-related disorders. Protein Misfolding Disorders are a recently recognized new group of diseases in which the hallmark event is a change in the secondary and/or tertiary structure of a normal protein without alteration of the primary structure.
The conformational change usually involves the reorganization of protein fragments adopting a-helical or random coil in the normal protein to a ß-sheet rich conformation in the pathological form. These changes may promote the disease by either a gain of a toxic activity or by the lack of biological function of the natively-folded protein. This group includes Alzheimer’s disease, transmissible spongiform encephalopathies (TSE) (also known as prion disorders), Huntington disease, serpin-deficiency disorders, hemolytic anemia, cystic fibrosis, diabetes type II, Amyotrophic Lateral Sclerosis, Parkinson disease, dialysis-related amyloidosis and more than 15 other less well-known diseases.
 Prion diseases (or TSE) are the prototype and best studied of all conformational disorders. TSE is a group of fatal neurodegenerative disorders affecting humans and other mammals and include Creutzfeldt-Jakob disease, Bovine Spongiform Encephalopathy and scrapie among others. The hallmark event is the misfolding and aggregation of an otherwise normal protein, named prion protein. In addition to have sporadic and inherited origins, TSE are also transmissible and the infectious agent (called prion) appears to be composed exclusively by the misfolded prion protein, which is able to transmit the disease to other individuals. During the process of prion replication the abnormally folded protein is capable to convert the normal protein into the misfolded one, inducing an exponential increase on the abnormal prion protein that most likely cause the disease by inducing neuronal apoptosis. Despite the fact that TSEs are relative rare diseases, they have gained significant attention from both the scientific community and society in general. This is due to the concerns generated by the recent appearance of a new human disease, variant Creutzfeldt-Jakob disease (vCJD), which has been linked to consumption of meat contaminated with bovine spongiform encephalopathy (BSE) and by the many unprecedented features, which have directly confronted popular dogmas in biology and makes the TSE field very attractive and challenging.
Prion diseases (or TSE) are the prototype and best studied of all conformational disorders. TSE is a group of fatal neurodegenerative disorders affecting humans and other mammals and include Creutzfeldt-Jakob disease, Bovine Spongiform Encephalopathy and scrapie among others. The hallmark event is the misfolding and aggregation of an otherwise normal protein, named prion protein. In addition to have sporadic and inherited origins, TSE are also transmissible and the infectious agent (called prion) appears to be composed exclusively by the misfolded prion protein, which is able to transmit the disease to other individuals. During the process of prion replication the abnormally folded protein is capable to convert the normal protein into the misfolded one, inducing an exponential increase on the abnormal prion protein that most likely cause the disease by inducing neuronal apoptosis. Despite the fact that TSEs are relative rare diseases, they have gained significant attention from both the scientific community and society in general. This is due to the concerns generated by the recent appearance of a new human disease, variant Creutzfeldt-Jakob disease (vCJD), which has been linked to consumption of meat contaminated with bovine spongiform encephalopathy (BSE) and by the many unprecedented features, which have directly confronted popular dogmas in biology and makes the TSE field very attractive and challenging.
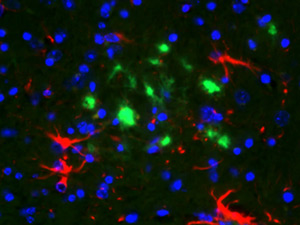 Alzheimer’s disease (AD) is the most common form of dementia in the late life, which at present does not have a cure or effective treatment. AD is a leading cause of death in the developed world and currently affect more than 10 million people worldwide. A hallmark event in AD is the misfolding of the amyloid-ß protein, which then became deposited in the brain in the form of amyloid plaques. These plaques are thought to cause neuronal death and synaptic loss resulting finally in dementia. Dr. Soto’s group developed a new model to explain amyloidogenesis in AD brains, which proposes that amyloid formation is triggered by conformational changes in the normal amyloid-ß protein. We also identified some of the factors that may induce the misfolding of the amyloid-ß protein and provided strong evidences that at least some of them (for example apolipoprotein E, RAGE receptor) might play a critical role in vivo. Based on the data generated by us and other groups, AD is now included in the group of disorders involving protein conformational changes as a key event in the pathogenesis.
Alzheimer’s disease (AD) is the most common form of dementia in the late life, which at present does not have a cure or effective treatment. AD is a leading cause of death in the developed world and currently affect more than 10 million people worldwide. A hallmark event in AD is the misfolding of the amyloid-ß protein, which then became deposited in the brain in the form of amyloid plaques. These plaques are thought to cause neuronal death and synaptic loss resulting finally in dementia. Dr. Soto’s group developed a new model to explain amyloidogenesis in AD brains, which proposes that amyloid formation is triggered by conformational changes in the normal amyloid-ß protein. We also identified some of the factors that may induce the misfolding of the amyloid-ß protein and provided strong evidences that at least some of them (for example apolipoprotein E, RAGE receptor) might play a critical role in vivo. Based on the data generated by us and other groups, AD is now included in the group of disorders involving protein conformational changes as a key event in the pathogenesis.
In the last eight years, the lab has been working on strategies for altering protein misfolding and aggregation in order to learn more about the molecular mechanism of this process and to generate novel approaches for therapy and diagnosis. The work has been built around two important discoveries that represents novel platform technologies: beta-sheet breakers peptides for preventing and correcting protein conformational changes and aggregation (therapeutic use) and the concept of cyclic amplification of protein misfolding for detecting the early pathogenic event in these diseases (diagnosis use).
Based on the knowledge of the structural determinants for protein misfolding, we have developed the concept of ß-sheet breaker peptides for the treatment of protein misfolding diseases. ß-sheet breaker peptides are short synthetic peptides homologous to the fragment of the protein undergoing misfolding, and engineered to contain residues that specifically block and reverse the conformational changes. ß-sheet breakers have been created to correct the misfolding of the amyloid-ß protein and the prion protein. The compounds have been demonstrated to be active in several in vitro and cellular models as well as in transgenic animal models for AD and in scrapie models of prion diseases. We have also characterized and improved the pharmacological properties of these compounds to make them suitable for in vivo use in CNS diseases. The first ß-sheet breaker peptide is currently under clinical evaluation in humans affected by AD. The same concept is now being expanded to generate ß-sheet breaker peptides aimed to correct the misfolding of other proteins implicated in some other diseases of the group of protein conformational disorders. Very promising results have been obtained, which allow us to propose that the concept of altering protein conformation by short synthetic peptides could form the basis of a novel therapeutic approach for a variety of diseases.
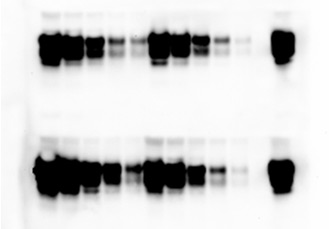 Cyclic amplification of protein misfolding (PMCA) is considered a major breakthrough in science and technology, because allows to mimic in vitro the pathological process associated to these diseases in a rapid and efficient way. The PMCA technology has been applied to convert large amounts of the normal prion protein into the abnormal form by incubating it with minute amounts of abnormal prion protein. The system consists on cycles of accelerated prion replication to reach an exponential increase in the conversion. These findings mark the first time in which the folding and biochemical properties of a protein have been cyclically amplified in a manner conceptually analogous to the amplification of DNA by PCR. PMCA might be helpful to understand the underlying biology of prions, to identify other factors that may be responsible for prion protein conversion, and to discover novel drug targets for prion diseases. In addition, PMCA has enormous potential in allowing current diagnostic tools to detect BSE and vCJD during the pre-symptomatic period and perhaps in living individuals, because it can multiply the number of prions facilitating their detection. Indeed, recent improvements of the technology has led to a more than 3 billion fold increase on sensitivity and the possibility to detect as few as 26 molecules of misfolded protein. This new developments demonstrate that PMCA is indeed working as PCR amplification. This level of sensitivity has recently enabled us to detect for the first time prions in the blood of sick as well as pre-symptomatic animals. These findings have had a major impact in various fields including the diagnosis of prion disease, blood banks safety, beef industry, etc. In addition, using the PMCA technology we have been recently able to generate infectious prion protein in vitro by propagating the protein misfolding process. Inoculation of wild-type animals with in vitro generated misfolded protein led to a disease with identical biochemical, histological and clinical features as transmissible spongiform encephalopathies. This experiment is widely considered by most scientists in the field as the final and definitive proof for the controversial prion hypothesis and as such is a major breakthrough in science.
Cyclic amplification of protein misfolding (PMCA) is considered a major breakthrough in science and technology, because allows to mimic in vitro the pathological process associated to these diseases in a rapid and efficient way. The PMCA technology has been applied to convert large amounts of the normal prion protein into the abnormal form by incubating it with minute amounts of abnormal prion protein. The system consists on cycles of accelerated prion replication to reach an exponential increase in the conversion. These findings mark the first time in which the folding and biochemical properties of a protein have been cyclically amplified in a manner conceptually analogous to the amplification of DNA by PCR. PMCA might be helpful to understand the underlying biology of prions, to identify other factors that may be responsible for prion protein conversion, and to discover novel drug targets for prion diseases. In addition, PMCA has enormous potential in allowing current diagnostic tools to detect BSE and vCJD during the pre-symptomatic period and perhaps in living individuals, because it can multiply the number of prions facilitating their detection. Indeed, recent improvements of the technology has led to a more than 3 billion fold increase on sensitivity and the possibility to detect as few as 26 molecules of misfolded protein. This new developments demonstrate that PMCA is indeed working as PCR amplification. This level of sensitivity has recently enabled us to detect for the first time prions in the blood of sick as well as pre-symptomatic animals. These findings have had a major impact in various fields including the diagnosis of prion disease, blood banks safety, beef industry, etc. In addition, using the PMCA technology we have been recently able to generate infectious prion protein in vitro by propagating the protein misfolding process. Inoculation of wild-type animals with in vitro generated misfolded protein led to a disease with identical biochemical, histological and clinical features as transmissible spongiform encephalopathies. This experiment is widely considered by most scientists in the field as the final and definitive proof for the controversial prion hypothesis and as such is a major breakthrough in science.
Our research has also focused on understanding the mechanism by which misfolded proteins induce cell death and tissue damage. Our recent work has demonstrated that misfolded prion protein induces neuronal apoptosis through endoplasmic reticulum (ER) stress pathway. Nano-molar concentrations of purified misfolded protein kill cells in culture by inducing the activation of the ER-resident caspase-12, which in turn activates the executor caspase-3. Interestingly, the initial response to ER-stress is a defense mechanism characterized by the up-regulation of ER chaperones. We have identified one of these chaperones as a key element on this pathway, which is specifically overexpressed in individuals affected by TSE and may lead to a novel strategy for early diagnosis.
Recently, we have become interested in the identification of non-disease-associated protein undergoing misfolding and aggregation as part of the normal biological function of the protein. Our recent studies show that a bacterial protein normally polymerizes as amyloid-like aggregates which participate in modulation the biological activity of the protein. The expansion of the amyloid and prion concepts towards many other proteins might revolutionize our understanding of biology.